Now loading...
Anthropic is collaborating with leading experts in synthetic, computational, and analytical chemistry to enhance Claude’s capabilities in this scientific field. A new white paper has been released, showcasing initial findings on Claude’s performance regarding NMR spectrum analysis, a fundamental task in chemistry.
Chemists often navigate a complex landscape of visual representations of molecules, including hand-drawn structures, database queries, and scientific notations. Each of these formats conveys the same chemical information but requires different skills to interpret. For instance, a hand sketch of caffeine can help identify its similarities to adenosine—a substance that signals drowsiness—enabling predictions about its effects. However, such sketches offer little assistance in distinguishing closely related molecules.
Identifying molecular structures accurately is essential, as chemistry plays an important role in everything from food and pharmaceuticals to consumer products. Even slight alterations in molecular structure can lead to entirely different substances, as seen with glucose and fructose, which share a formula but are metabolized differently. Similarly, converting a molecule into its mirror image can transform a sedative into a harmful teratogen, emphasizing the necessity for precision in identifying molecules in various contexts.
Translating these molecular representations can be a time-consuming challenge, particularly in the face of an expanding chemical database. The largest chemistry registry, CAS, currently includes over 290 million compounds and adds around 15,000 new entries every day.
While artificial intelligence has the potential to alleviate these research burdens, its application in chemistry has remained largely theoretical until recently. Machine learning tools have been touted as revolutionary for tasks such as retrosynthesis planning and reaction prediction, but the inconsistent quality and accessibility of data have hindered their practical use. Even with capable AI models available, adoption remains sporadic, particularly among academic and smaller lab chemists.
Recent developments, however, indicate that AI can now tackle challenging chemical problems. The latest multimodal models can directly interpret chemical structures from various sources and read experimental details as published, rather than relying on structured databases. These models also provide step-by-step reasoning that allows chemists to verify the output, even if the persistent data issues continue to influence their efficacy.
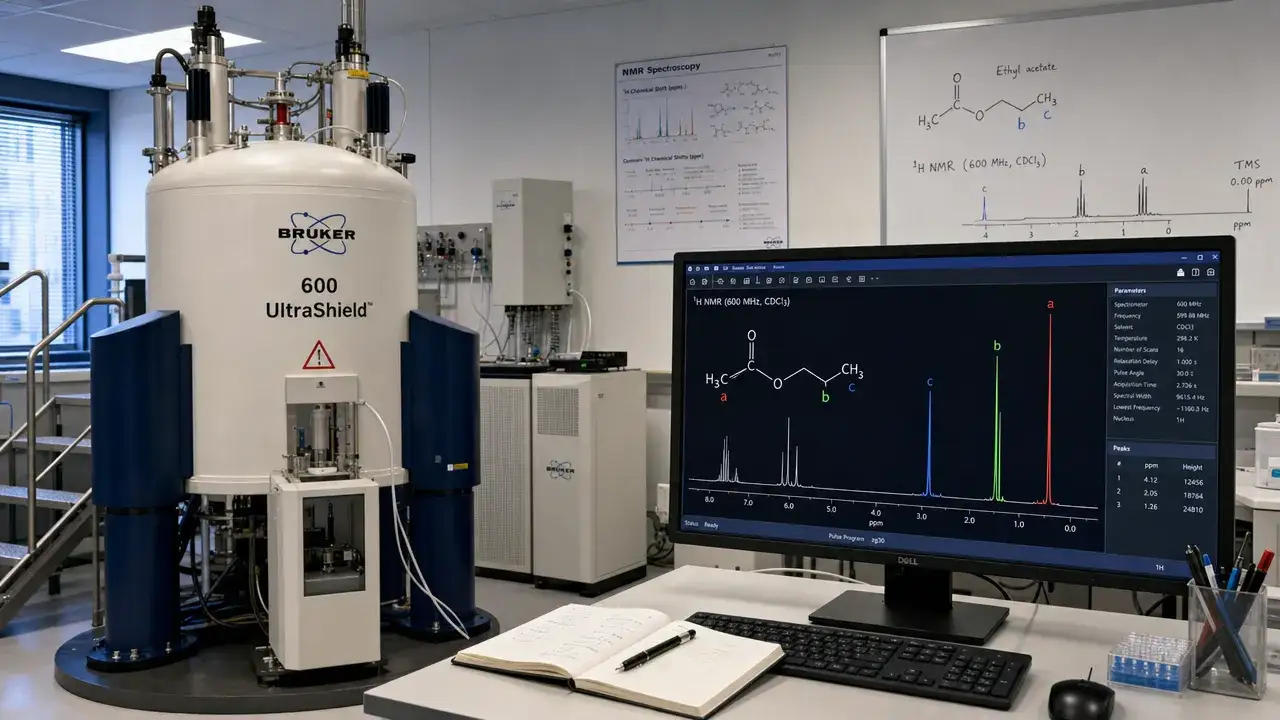
This assessment of Claude’s capabilities indicates that it can effectively assist chemists with the repetitive tasks of translating and integrating complex data. The recent study focuses on NMR (nuclear magnetic resonance) spectroscopy, a cornerstone technique that enables chemists to derive molecular structure from spectral data.
The research compared Claude’s performance against prominent software solutions like ChemDraw and MestReNova across 20 compounds recently published in synthetic chemistry preprints. The evaluation involved both forward predictions—simulating the NMR spectrum from a given structure—and inverse predictions, wherein Claude attempted to deduce the structure from the experimental spectrum, a significantly more challenging task.
In evaluating the predictive accuracy of each tool, Claude outperformed expectations. The study found that Claude’s Opus 4.7 model achieved the highest accuracy in predicting hydrogen peaks, landing with an average error significantly below acceptable thresholds. On carbon peaks, Opus 4.7 and MestReNova performed comparably, showcasing both tools’ relevance in real-world applications.
The evaluation also highlighted Opus 4.7’s ability to consistently predict sub-peak spacing and maintain accuracy across multiple runs. In subsequent tests, Claude was remarkably successful in deducing structures from spectral data, recovering all simpler structures correctly and achieving strong performance on more complicated targets when provided with additional contextual information.
Overall, the findings indicate that a general-purpose AI model like Claude can match or even exceed the performance of established NMR software solutions on average, including its capacity to perform inverse elucidation tasks. Traditional software for structure elucidation has existed for years but often relies on complex setups and specialized training; Claude provides a more accessible alternative by using the same input types a chemist would typically use.
The study does recognize several limitations, including the small sample size and constraints on the types of chemical structures tested. Future research is planned to overcome these gaps by examining additional compound classes and solvents, as well as enhancing Claude’s understanding of chemical literature.
As Anthropic continues to refine Claude’s chemistry capabilities, the focus will remain on addressing specific challenges that slow chemists’ workflows, ranging from interpreting chemical structures to understanding the nuances of chemical literature. By improving performance in these areas, Anthropic hopes to provide tools that genuinely assist chemists in their research efforts.
Researchers interested in collaborating on projects where Claude could be of assistance are encouraged to reach out to the AI for Science program.
